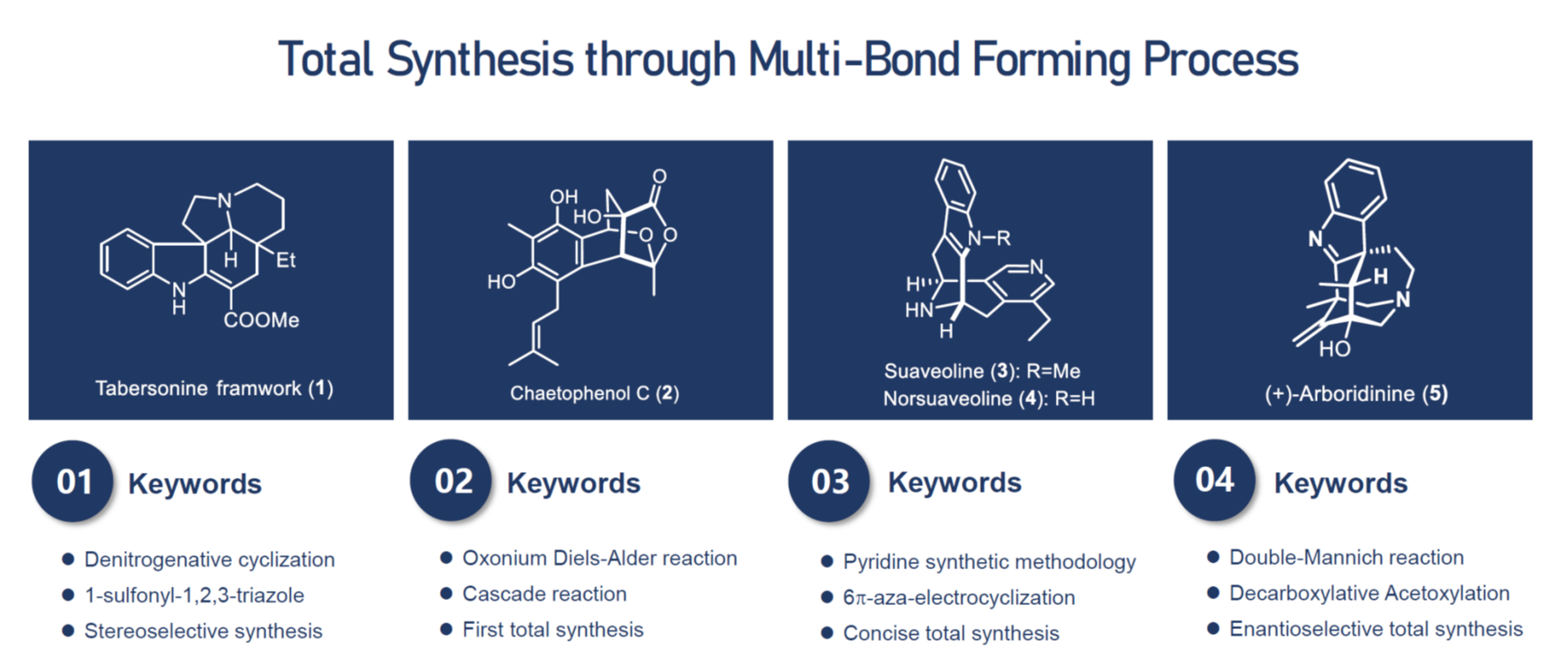
Synthesis of Tabersonine framwork
We have developed a rhodium-catalyzed intramolecular [3 + 2] cycloaddition with N-sulfonyl-1,2,3-triazoles as the 1,3-dipole precursor, which showed excellent stereoselectivity and allowed for a quick access to the scaffold of Aspidosperma- and Kopsia-related alkaloids. Based upon its operational simplicity and the mild reaction conditions, the current approach may open up a new and efficient access to an array of valuable indole alkaloid analogues. The total synthesis towards the related alkaloids is ongoing.

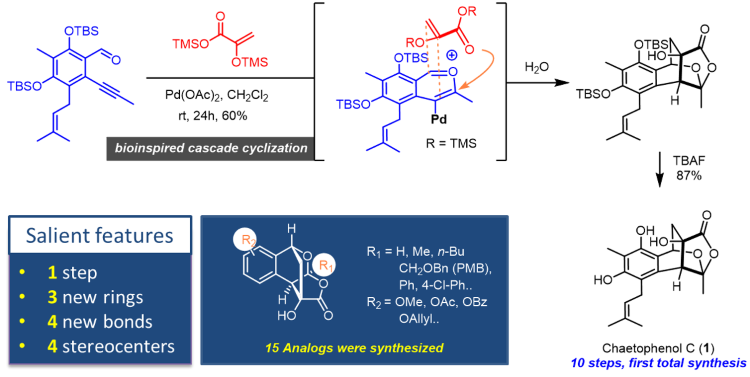
First total synthesis of Chaetophenol C
We developed a palladium-catalyzed cascade cycloaddition/cyclization reaction of ortho-alkynylbenzaldehydes giving products containing the [2.2.2] bicyclic lactone core structural unit found in chaetophelnol C which was isolated in 2013 from epigenetic manipulated Chaetomium indicum by Oshima and co-workers. The new reaction not only leading to a one-step construction of tetracyclic core structure of chaetophenol C but also providing its correctly placed functionality from two simple starting materials under very mild condition. The developed chemistry was also successfully applied to the first total synthesis of chaetophenol C and dozens of its analogs.
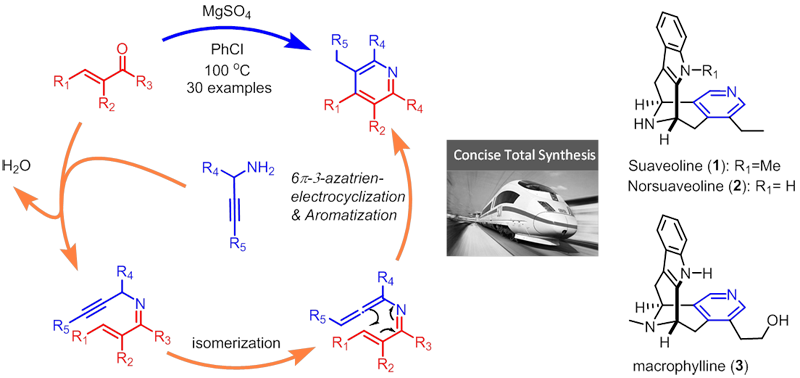
Total synthesis of pyridine containing Suaveoline natural products
We have developed a new, metal-free, and one-pot synthetic route to substituted pyridines, which employs a cascade reaction sequence from an α,β-unsaturated ketone and a phosphazene (both produced in situ), involving an aza-Wittig, a 6π-3-azatriene electrocyclization reaction, and a [1,3]-hydrogen shift. Based upon advantages such as simple operation, broad functionality tolerance, and significance of the multisubstituted pyridine products, this method provide a significant potential in organic synthesis. Recently, the total synthesis of three pyridine-containing natural products including suaveoline, norsuaveoline and normacrophyline was achieved in our lab by using our own-developed methodology.
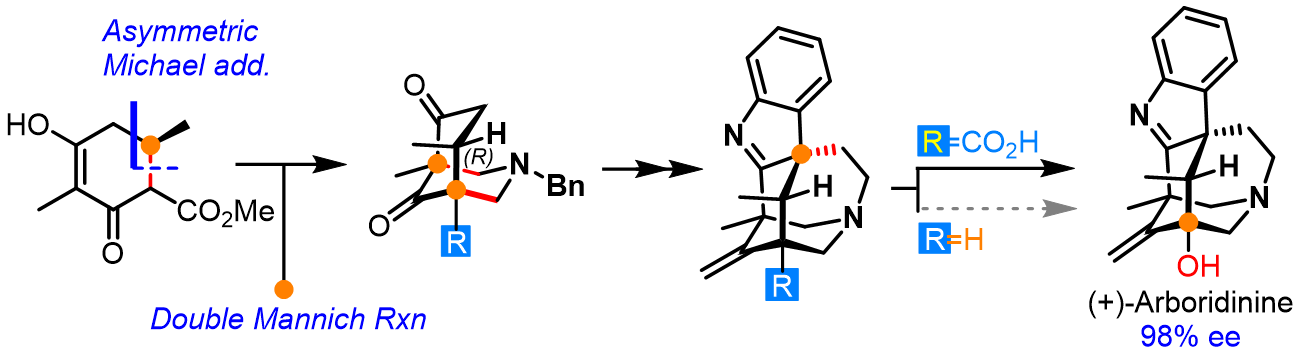
Enantioselective total synthesis of (+)-Arboridinine
The enantioselective total synthesis of cage-shape alkaloid (+)-arboridinine is reported. The synthesis takes advantage of stereoselective double-Mannich reaction to rapidly construct the aza[3.3.1]bicyclic core along with two quaternary stereocenters of the alkaloid. Key steps for the present synthesis include an enantioselective Michael addition establishing the original chiral center at C10, and intramolec-ular dearomative alkylation forging the cage-shape ring system found in arboridinine, as well as creating the requisite quaternary carbon center at C3. The bridgehead hydroxyl moiety at C11 was installed through a late-stage cobalt-catalyzed decarboxylative acetoxylation reaction. This strategy enables a 14-step asymmetric total synthesis of (+)-arboridinine from the readily available starting materials with most of the transformations performed on the decagram scale.